

Back to top
miCORE Whole Genome Metagenomics

Unbiased community profiling. Reveal taxonomy and function at species- or strain-level precision. Transcend domain boundaries for a truly comprehensive view.
From soil and water to host-associated samples, shotgun metagenomics enables comprehensive analysis of all DNA in your sample. Go beyond marker genes to uncover taxonomic composition, metabolic potential, and functional elements such as antibiotic resistance and virulence factors.
From soil and water to host-associated samples, shotgun metagenomics enables comprehensive analysis of all DNA in your sample. Go beyond marker genes to uncover taxonomic composition, metabolic potential, and functional elements such as antibiotic resistance and virulence factors.
What You Can Achieve
- Characterize bacterial and fungal communities
- Identify organisms and estimate relative abundances down to species or strain level
- Detect viruses in complex samples, including host tissues and exudates
- Explore metabolic functions, e.g., antibiotic resistance genes,virulence factors, etc.
- Conduct gene-level analysis of uncultivable microorganisms
- Track community shifts and evolutionary adaptations under changing conditions
Before You Start
To set up your shotgun metagenomics project effectively, consider:
- Sample source and quantity – origin and amount of material available
- Sequencing depth – level of sensitivity needed to detect rare organisms
- Community complexity – taxonomic diversity and expected composition
- Research focus – taxonomy, function, or both
- Method choice – is Whole Genome Metagenomics or Amplicon Metagenomics the better fit?
- Data analysis – availability of computational resources and bioinformatics expertise
Our experts support you from experimental design to bioinformatics analysis.
Modular Workflow
Outsource the complete workflow or select specific modules. Our shotgun metagenomics process is designed for flexibility.
Typical workflow steps include:

Bioinformatics Analysis
Our Whole Genome Metagenomics analysis module combines intensive computing with optimized tools for reliable results. Typical analyses include:
- Data quality control
- Taxonomic profiling of organisms from all biological domains
- Clustering of samples based on community composition
- Functional annotations
- Visualization with interactive charts (e.g., Krona plots)
Example Results
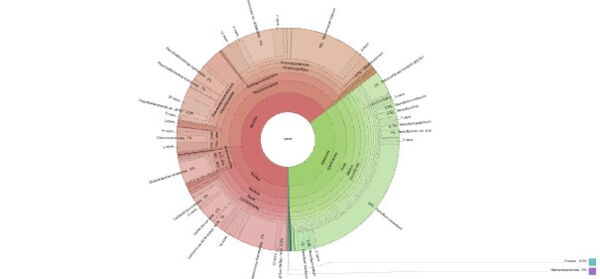
[left] Click to explore the interactive Krona chart showing taxonomic composition from domain to species level.





Turnaround Time
- Sequencing only: 20 working days
- Full analysis: +15 working days
- Taxonomy-only analysis: +5 working days
- Express service available upon request
Sample Requirements
- Buffer recommendation: 10 mM Tris-HCl (pH 7.5–8.5)
- Avoid: EDTA >1 mM
- DNA quantification: Fluorometric methods (e.g., PicoGreen®, Qubit®)
- Please contact us for project-specific input on DNA amount and quality
Notes
- Guidance and customized workflows and additional downstream analyses are available on request. Please use the contact form on the right.
- Related services: miCORE Amplicon Metagenomics, nanoCORE Amplicon Metagenomics, Shotgun Metatranscriptomics, BacterialSeq.