

Back to top
Séquençage de Novo

- Assembler le génome d'une espèce génétiquement non caractérisée
- Détecter les gènes non découverts
Aperçu
Considérations avant de lancer un projet de séquençage de novo :
- Qualité de l'ADN et contaminations potentielles ?
- Illumina ou PacBio ?
- Longueur des reads ?
- Taille de la bibliothèque ?
- Profondeur de séquençage ?
- Voisinage taxonomique ?
- Études de suivi ?
Laissez-nous vous guider - de la conception à l'analyse
Exemples de projets utilisant le séquençage de novo :
- Assemblage à partir du séquençage de novo de bactéries halophiles non caractérisées
- Assemblage de novo d'un champignon connu mais fortement muté
- Construire un projet de génome de référence pour une plante agricole importante
- Assemblage de reads non cartographiés dans le cadre d'un projet de reséquençage (nouveaux plasmides, grandes insertions)
- Séquençage bactérien de novo fréquent
Les demandes relatives au séquençage de novo :
- Génération d'un transcriptome de référence
- Reséquançage
Déroulement

Pour plus d'informations et une description technique détaillée, veuillez télécharger notre note d'application "Séquençage Illumina de novo" (voir les téléchargements correspondants).
Résultats
L'objectif principal d'un assemblage est d'aligner des reads uniques du séquençage NGS ou Sanger dans des contigs, plus grands, et dans des séquences génomique entières. L'assemblage permet de comprendre la structure génomique d'un organisme et l'organisation de son contenu génétique.
Notre module d'assemblage peut traiter des reads provenant de toutes sortes d'organismes, y compris les génomes bactériens et les organismes eucaryotes plus complexes. Deux approches sont possibles: assemblage de novo ou alignement avec référence.
Les résultats du module sont des contigs assemblés ainsi que des données statistiques qui répondent aux questions suivantes :
- Quelle est la longueur totale cumulée des contigs assemblés ? (voir figure 1)
- Quel est le contenu en GC des contigs assemblés ? (voir figure 2)
- Quelle est la répartition par taille des contigs assemblés ? (voir figure 3)
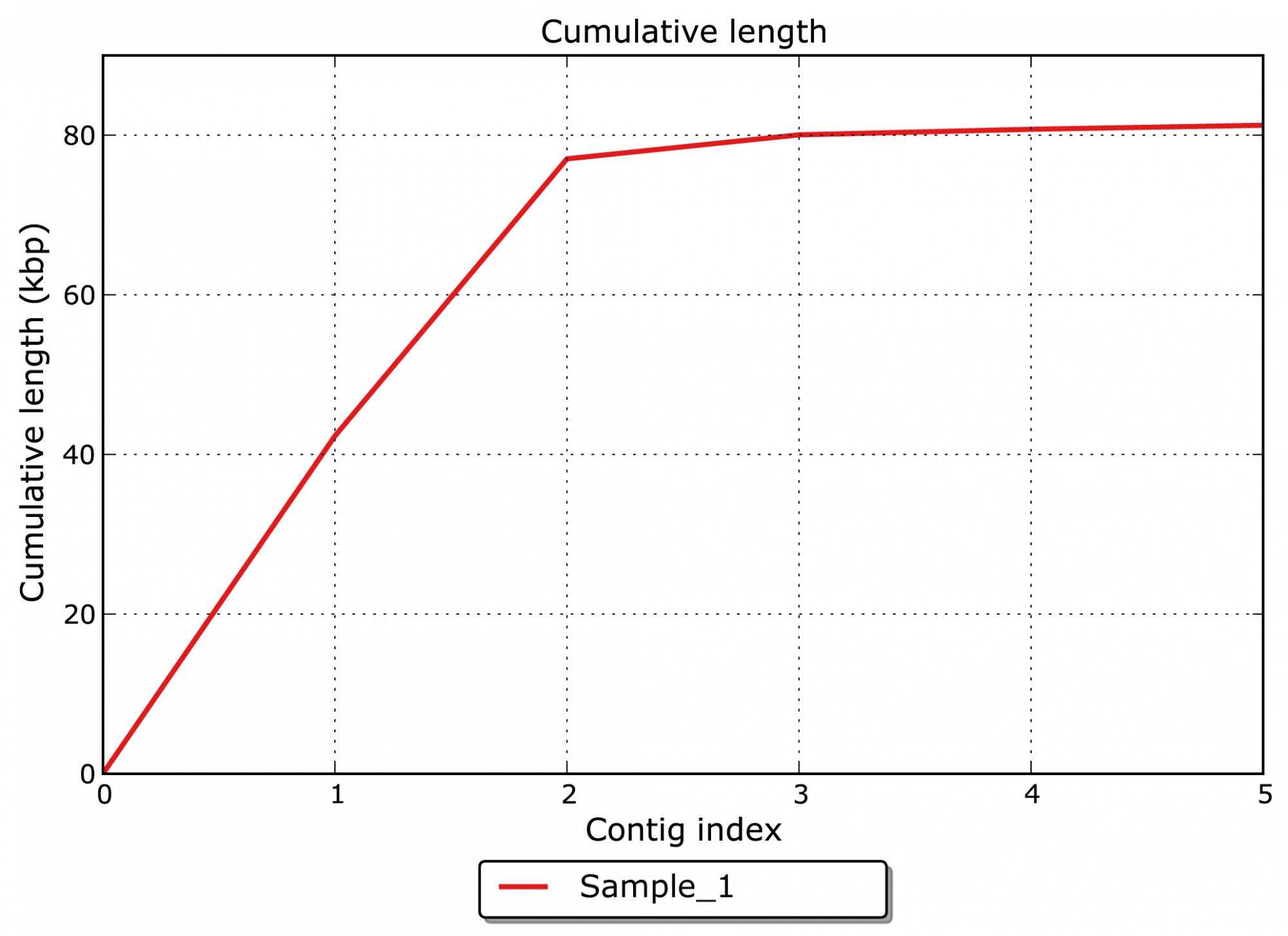
Figure 1: Longueur cumulée de l'assemblage.
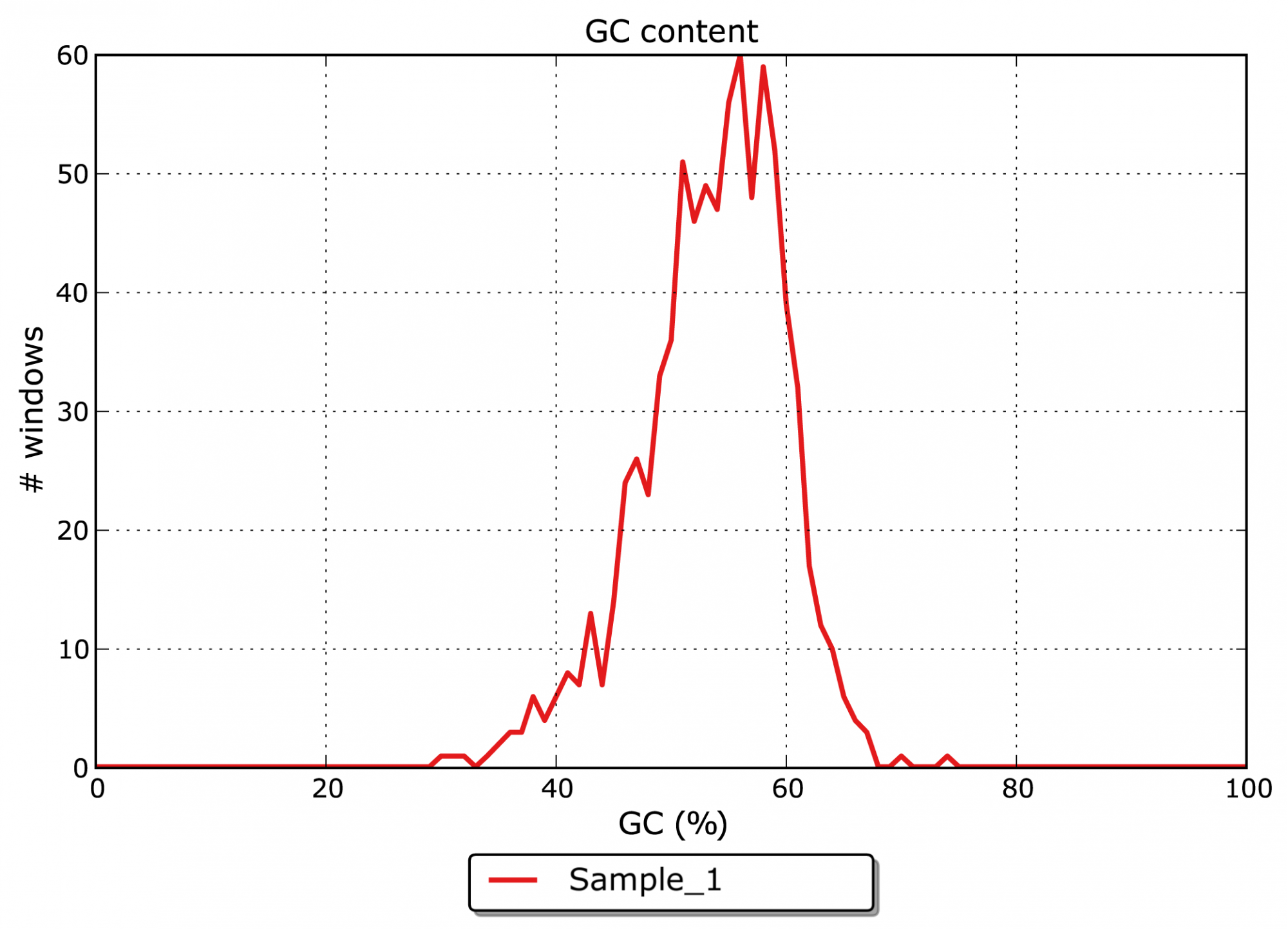
Figure 2: Distribution du GC % pour les contigs assemblés dans les fenêtres de 100 paires de bases sans chevauchement.
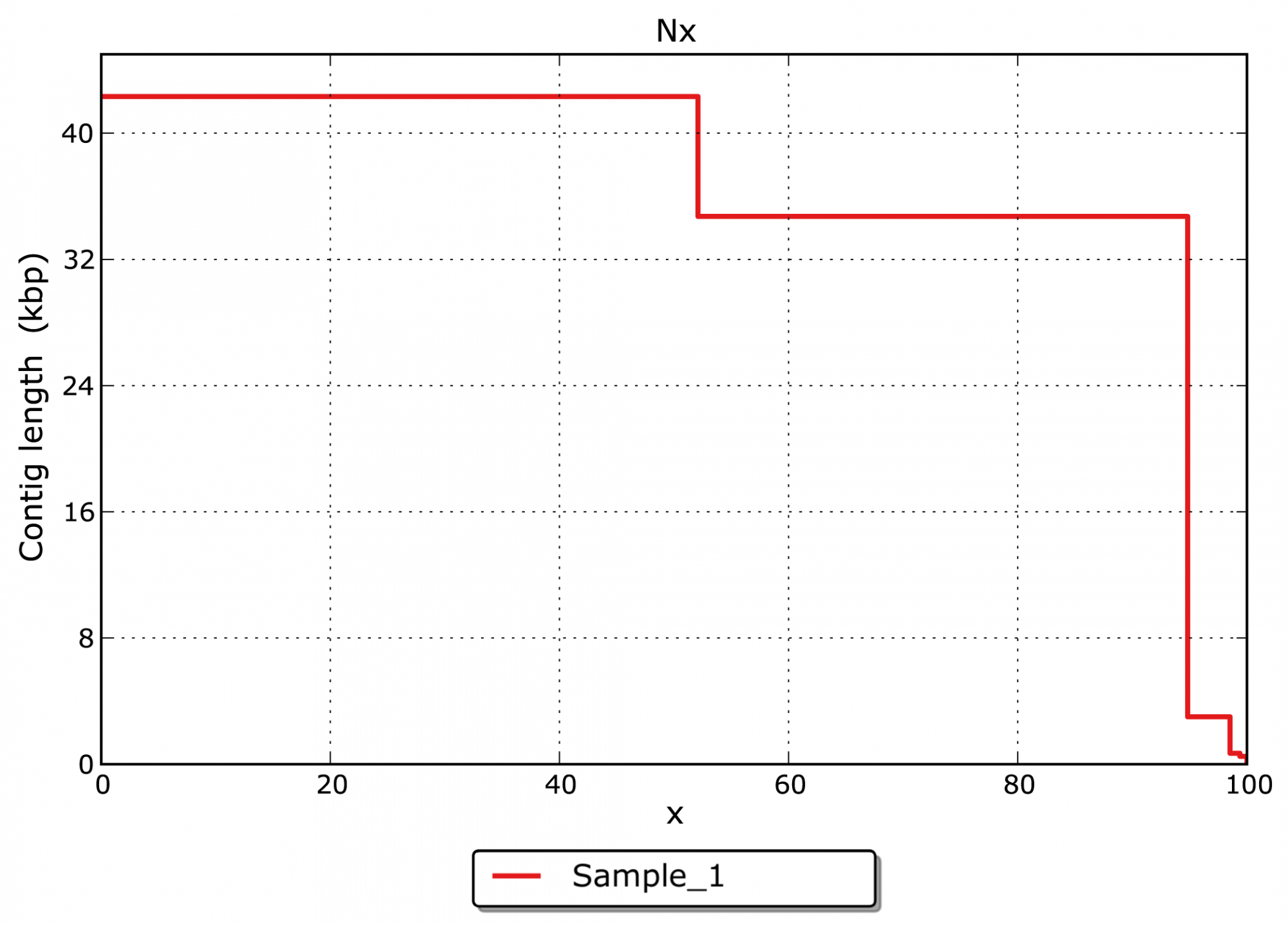
Figure 3: Statistiques Nx pour les tailles de contig.
Délai d'exécution
- Livraison des données dans les 20 jours ouvrables suivant la réception de l'échantillon (y compris la préparation de la bibliothèque et le séquençage)
- 15 jours ouvrables supplémentaires pour l'analyse des données (bioinformatique)
- Service express possible sur demande