

Back to top
miCORE Amplicon Metagenomics

- Explorer la composition taxonomique de la communauté - bactéries ou champignons - de vos échantillons
- Comparer les changements taxonomiques dans un cadre expérimental
- Optimisez votre prochaine expérience d'échantillonnage
Aperçu
Considérations avant de lancer un projet de métagénomique des amplicons :
- Objectif scientifique
- Matériel de départ et services souhaités (de l'isolement à la bioinformatique)
- Résultat : enquête, expérience, potentiel fonctionnel
Laissez-nous vous guider - de la conception à l'analyse
Exemples de projets utilisant la métagénomique des amplicons :
- Détecter les changements de la communauté bactérienne intestinale en fonction de la nutrition
- Étudier la diversité génétique des archaebactéries dans les environnements de cheminées hydrothermales en eaux profondes
- Observer les changements de communauté dans un bioréacteur
- Caractériser les spores fongiques de l'air
Applications liées à la métagénomique des amplicons :
- Amplicon Métagénomique personnalisé
- Métagénomique du génome entier
- Métatransciptomique
Déroulement

Pour plus d'informations et une description technique détaillée, veuillez télécharger notre application "miCORE Amplicon Metagenomics" (voir les téléchargements correspondants).
Résultats
Sans Bioinformatique
Données brutes
Si vous n'avez pas commandé de module d'analyse bioinformatique, Microsynth fournit les résultats clés suivants pour la métagénomique 16S/ITS :
- Évaluation de la quantité et de la qualité du séquençage (au format .xlsx)
Une évaluation de la quantité et de la qualité de vos données de séquençage. - Données brutes (par échantillon, au format .fastq)
Les données brutes vous permettent d'effectuer vos propres analyses ou de remonter jusqu'à chaque nucléotide séquencé. - Rapport de synthèse du projet (format .pdf)
Un rapport résumant les paramètres clés du projet.
Avec Bioinformatique
Analyse bioinformatique standard
Avec le module d'analyse bioinformatique standard de Microsynth pour la métagénomique 16S/ITS, vous obtiendrez des informations complètes :
- Rapport détaillé (en format .html interactif)
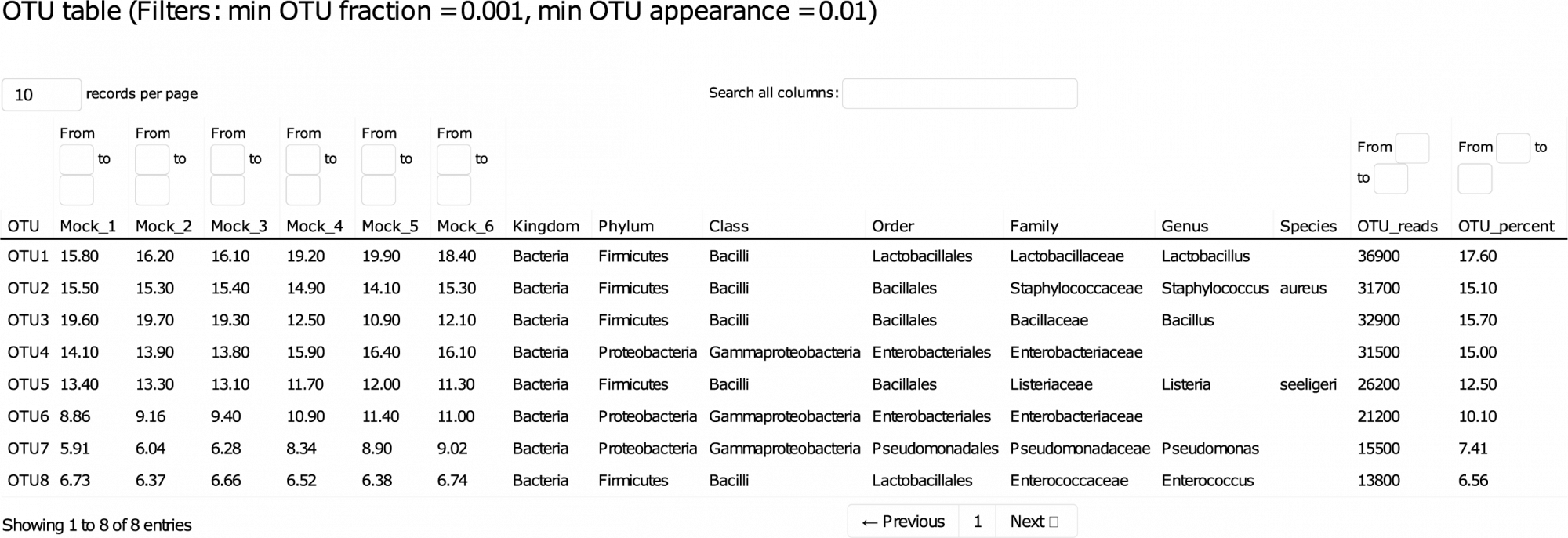
Ce rapport détaillé vous guide à travers les données, permettant le tri et le filtrage des résultats clés de manière interactive. - Variants de séquence d'amplicon (ASV) (au format .fasta, voir tableau 1)
ASVs, également connus sous le nom d'unités taxonomiques opérationnelles à rayon zéro (zOTUs), au format .fasta. - Abondance des ASV, taxonomie et confiance dans les prédictions (en format .tsv et .xlsx)
Aperçu de l'abondance des ASV, de la taxonomie et de la confiance dans les prédictions. - Analyse de la diversité intra-échantillon et courbes de raréfaction (en format .tsv et .pdf, voir Figure 1)
Explorer la diversité intra-échantillon et estimer la couverture métagénomique à l'aide de courbes de raréfaction. - Visualisation du métagénome via les graphiques Krona (en format .html interactif, voir Figure 2)
Interprétez et explorez facilement vos données métagénomiques grâce aux graphiques interactifs de Krona.
Ces résultats fournissent une compréhension complète de votre expérience, d'une vue d'ensemble jusqu'au niveau des nucléotides, ce qui vous permet de prendre des décisions éclairées et de tirer des conclusions significatives.
Analyse bioinformatique complémentaire (moyennant des frais supplémentaires) :
1. Analyse comparative
- Diversité inter-échantillons (en format .tsv et .pdf, voir figure 3)
Obtenez des informations sur la diversité entre les échantillons, présentées sous forme de tableaux et de graphiques. - Analyse en composantes principales (en format .tsv et .pdf, voir figure 3)
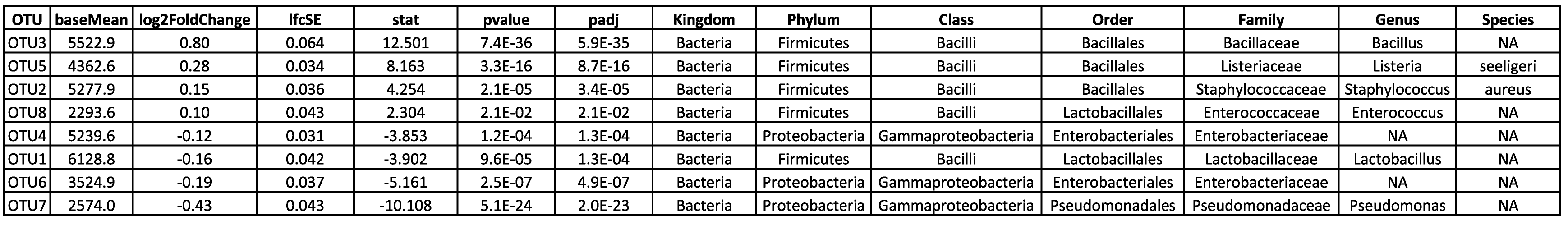
Visualisez et interprétez des relations complexes grâce à l'analyse en composantes principales. - Analyse des ASV différentiels (en format .tsv et .html, voir tableau 2)
Identifiez et explorez les ASV différentielles grâce à des résultats d'analyse détaillés disponibles sous forme de tableaux et de fichiers HTML. - Visualisation de l'analyse statistique
Accédez à des représentations visuellement informatives des analyses statistiques.
Ces services supplémentaires permettent d'approfondir la compréhension et l'interprétation, améliorant ainsi votre capacité à extraire des informations précieuses de vos échantillons.
2. Profilage fonctionnel
- Inférence et prédiction des profils fonctionnels (format .tsv et .html, voir tableau 3)
Prédire le potentiel génétique fonctionnel de vos échantillons sur la base des enquêtes et des informations génétiques complètes des taxons identifiés.
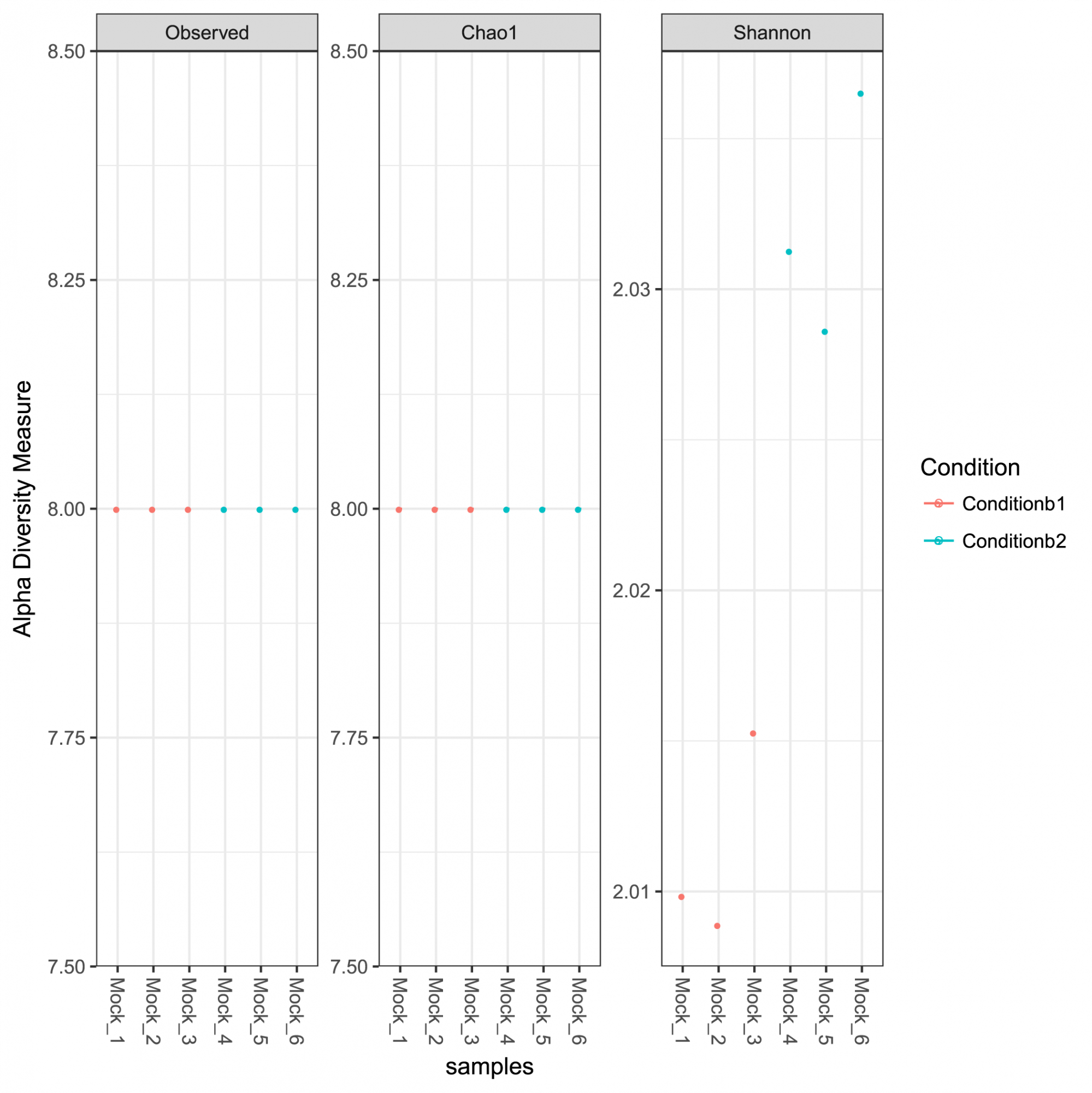
Figure 1: Mesures de la diversité alpha pour la communauté analysée, y compris la richesse observée, les indices Chao 1 représentant la richesse estimée et les indices de diversité Shannon.
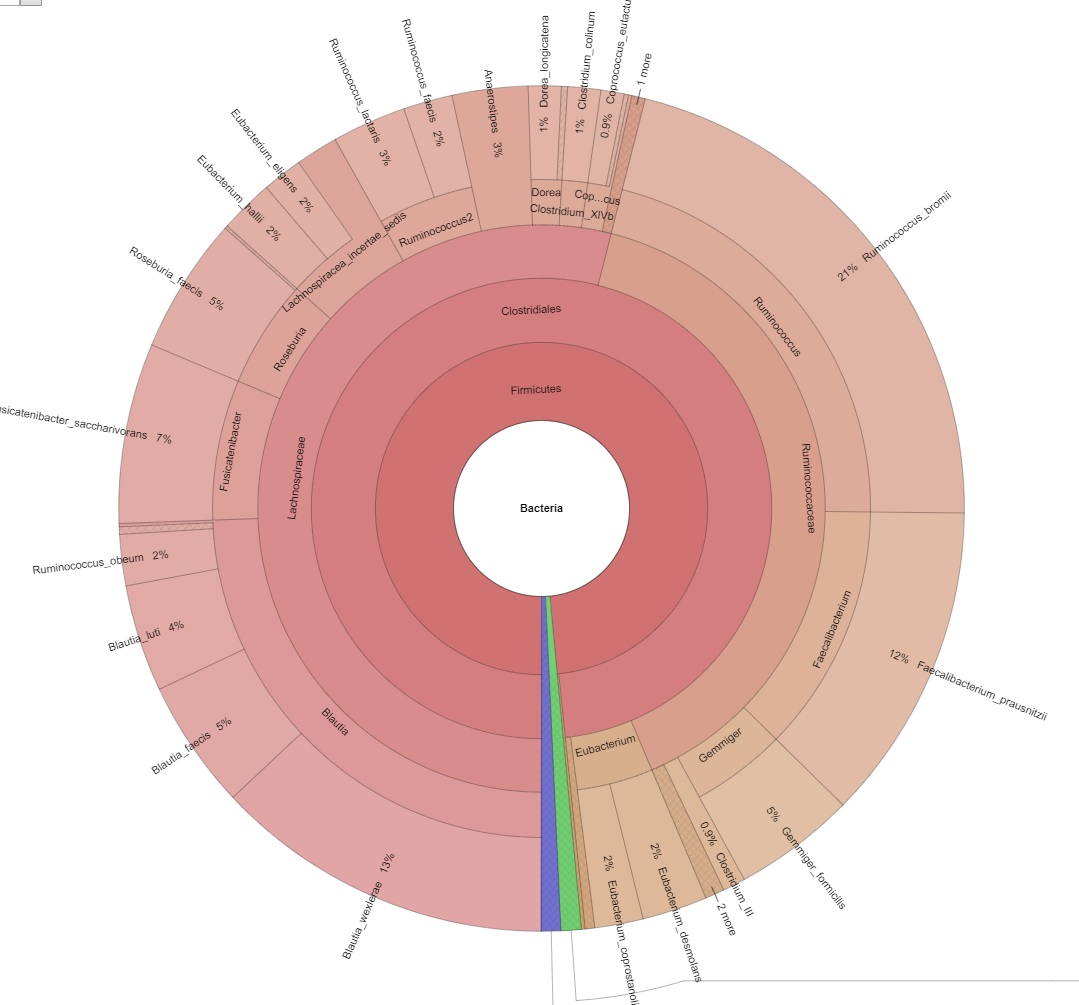
Figure 2 : Image d'un graphique interactif de Krona.
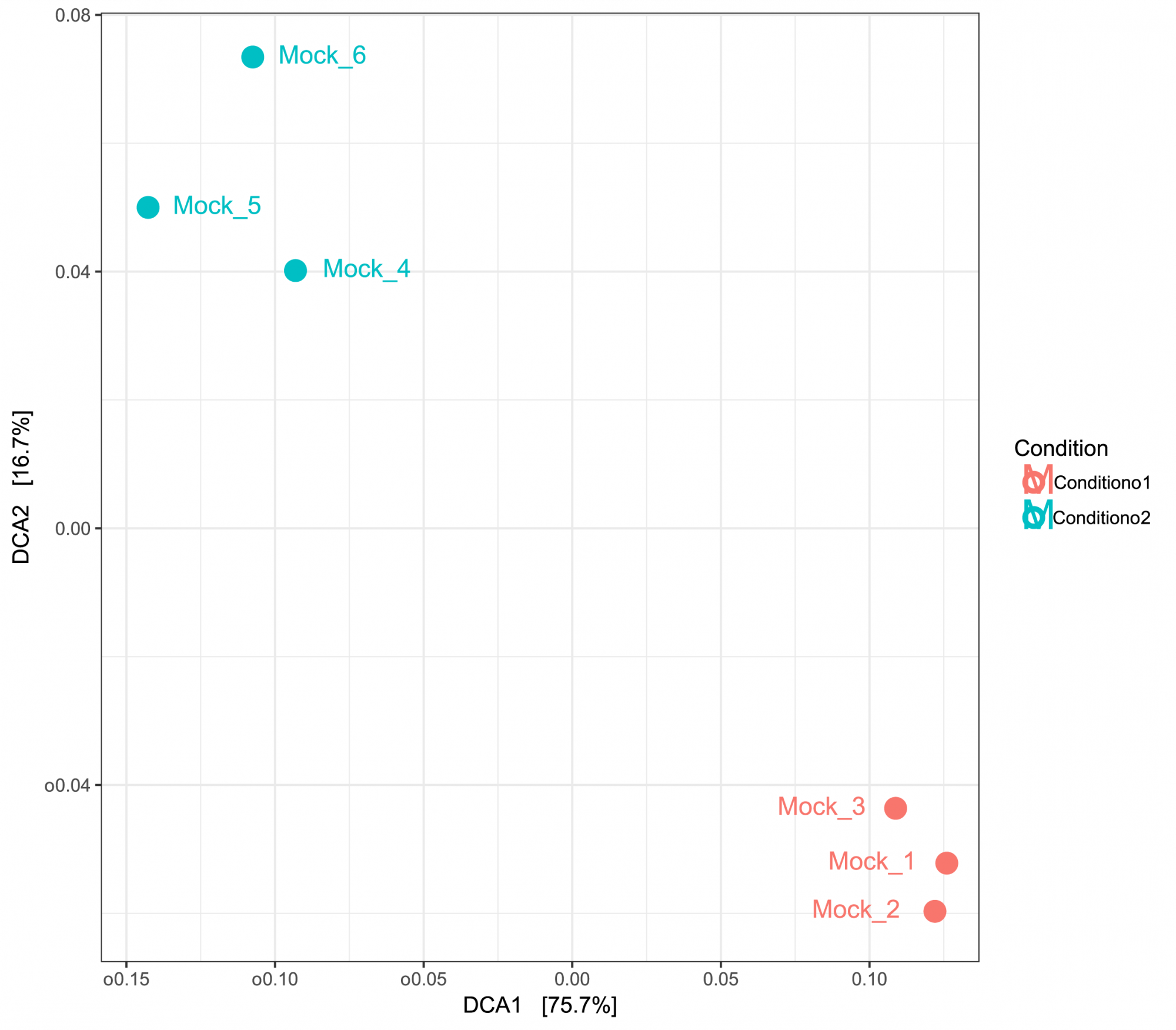
Figure 3: ACP basée sur les distances UniFrac montrant des similitudes entre les échantillons. Les échantillons ont été regroupés en fonction de deux conditions.

Délai d'exécution
- Livraison des données dans les 15 jours ouvrables suivant la réception de l'échantillon (y compris la préparation de la bibliothèque et le séquençage)
- 5 jours supplémentaires pour l'isolement de l'ADN et 3 jours pour l'analyse des données (bioinformatique)
- Service express possible sur demande