

Back to top
miCORE Resequencing

- Détecter les mutations héritées ou acquises au sein d'un génome.
- Étudier les comparaisons à l'échelle du génome.
- Vérifier la souche ou le type de cellule au niveau du génome.
- Détecter les mutations hors cible de votre expérience d'édition de gènes CRISPR/Cas9.
Aperçu
Considérations avant de lancer un projet de séquençage du génome entier :
- Objectif scientifique
- Qualité de la génome de référence
- Détection de SNV (Variant Calling)
- Couverture optimale/profondeur de séquençage et longueur de lecture
- Des contaminations substantielles de l'ADN sont-elles suspectées ?
Laissez-nous vous guider - de la conception à l'analyse
Exemples de projets utilisant le séquençage du génome entier :
- Détection des mutations acquises par les cellules cancéreuses - des SNP aux grandes variations structurelles
- Détection du site d'insertion
- Vérification des mutations, excluant les mutations hors cible
- Modifications génétiques dans les études de sélection
Applications liées au séquençage du génome entier :
- RNA Sequencing
- Whole Exome Resequencing
- De Novo Sequencing
Déroulement

Résultats
Sans Bioinformatique
Données brutes :
Si aucun module d'analyse n'est commandé, Microsynth fournit pour le séquençage du génome entier les résultats clés énumérés ci-dessous :
- Évaluation de la quantité et de la qualité du séquençage (au format .xlsx)
L'évaluation de la quantité et de la qualité des données de séquençage. - Données brutes (par échantillon, au format .fastq)
Les données brutes vous permettent d'effectuer vos propres analyses ou de retrouver chaque nucléotide séquencé. - Un rapport de synthèse du projet (format .pdf)
Ce rapport résume les principaux paramètres du projet.
Avec Bioinformatique
Analyse bioinformatique standard :
Pour notre application de reséquençage, en plus des données brutes, le module d'analyse de Microsynth fournit une variété d'informations pour répondre à vos objectifs scientifiques :
- Évaluation de la quantité et de la qualité du séquençage (en format .xlsx et .html)
Une évaluation complète de la quantité et de la qualité du séquençage, fournissant des informations cruciales. - Fichiers d'alignement/de cartographie et index (au format .bam et .bai)
Accédez aux fichiers d'alignement et de cartographie ainsi qu'aux index correspondants pour plus de commodité. - Analyse de la couverture du génome et de la profondeur de lecture (au format .tsv et .bed ; voir figure 1)
Obtenez un aperçu de la couverture du génome et de la profondeur de lecture grâce aux résultats fournis aux formats .tsv et .bed. - Variations structurelles & Analyse du nombre de copies (comparer le tableau 1 & la figure 2)
- Appel de variants SNV et de petits InDels (au format .vcf, voir tableau 2 et tableau 3)
Identifier les variants de nucléotides simples (SNV) et les petites insertions/délétions (InDels < 50 pb) grâce aux résultats de la recherche de variants disponibles au format .vcf. - Annotation des variations ayant des conséquences au niveau des acides aminés (au format .html, voir tableau 4)
Comprendre les variations ayant des conséquences potentielles au niveau des acides aminés, présentées au format .html (accessibles pour la plupart des organismes modèles/si le génome de référence est annoté en conséquence).
Facultatif :
- Filtrage des variantes par rapport à un échantillon de fond (en format .html)
Personnalisez votre analyse en filtrant les variants par rapport à un échantillon de fond, les résultats étant fournis au format .html. - Séquence consensus de l'échantillon (en format .fasta et .gb)
Recevez la séquence consensus d'un échantillon, qui peut donner lieu à une séquence chimérique, aux formats .fasta et .gb.
Analyse bioinformatique complémentaire (moyennant un coût supplémentaire)
Épidémiologie génomique (pour les procaryotes uniquement) :
- Recherche de correspondances avec les bases de données de résistance et de virulence et les gènes de mycotoxines (en format .tsv)
Identification des correspondances avec les bases de données sur la résistance et la virulence, les résultats du criblage étant fournis au format .tsv. - Typage phylogénétique de la souche de l'échantillon (au format .pdf)
Comprenez la relation phylogénétique de la souche de votre échantillon grâce à un rapport concis au format .pdf.
Ces résultats vous donnent une vue d'ensemble de votre analyse de reséquençage, vous permettant d'extraire des informations significatives et de prendre des décisions éclairées.

Tableau 1: Détail d'un tableau de résultats montrant les variations de nucléotides uniques détectées, les petites insertions et délétions et leur annotation.
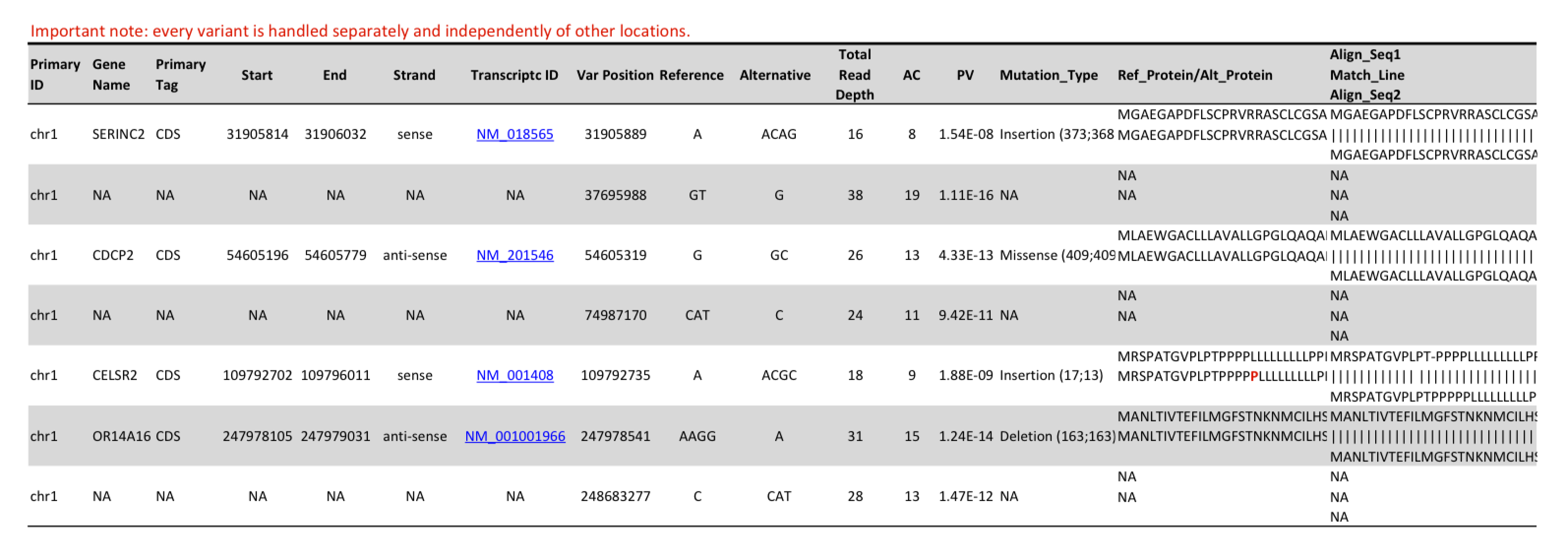

Tableau 3: Détail d'un tableau de résultats énumérant les variations structurelles putatives.
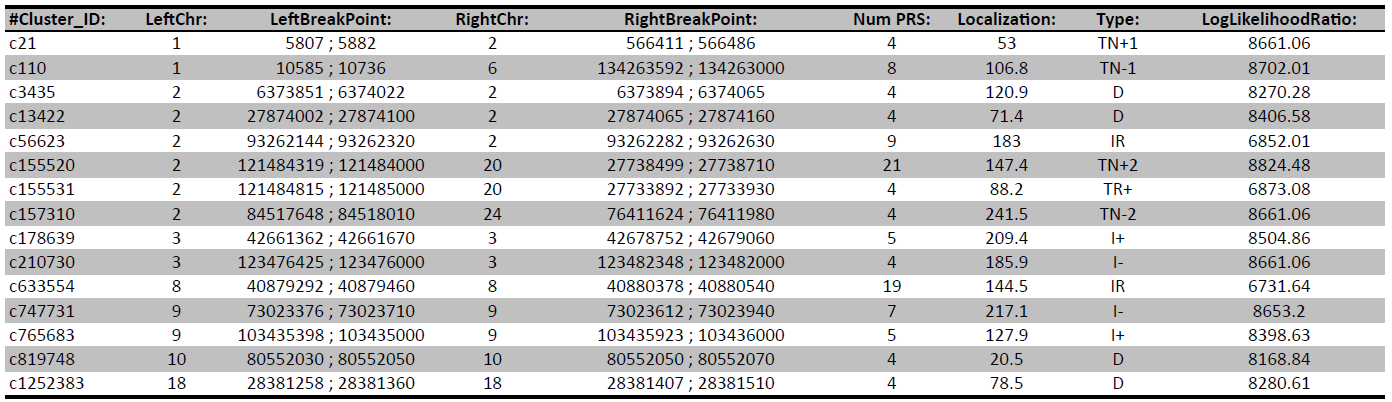
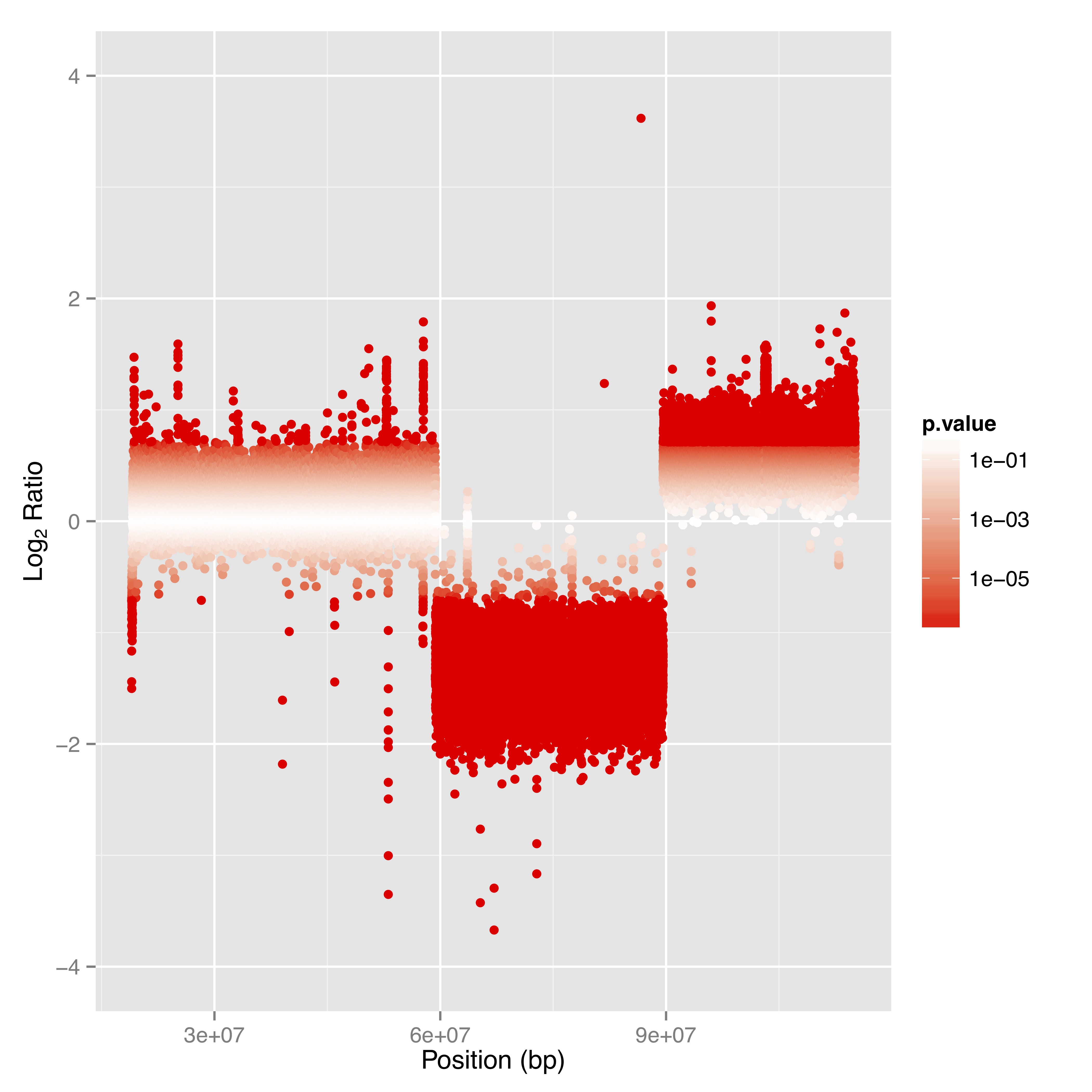
Figure 2: Ce graphique représente une variation possible du nombre de copies par rapport à un échantillon de référence.
Délai d'exécution
- Livraison des données dans un délai de 20 jours ouvrables à compter de la réception de l'échantillon (y compris la préparation de la bibliothèque et le séquençage).
- 3 ou 6 jours ouvrables supplémentaires pour l'analyse des données (bioinformatique ; petits ou grands génomes).
- Service express possible sur demande.