

Back to top
Genomische Epidemiologie

Verwenden Sie den Genomische Epidemiologie Analyse Service von Microsynth, um:
- Genome de novo zu assemblieren und zu annotieren
- auf das Vorhandensein von Virulenz-, Resistenz- und Mykotoxin-Genen zu prüfen
- mehrere Bakterienstämme gleichzeitig zu typisieren (z. B. cgMLST-Analyse)
- die Zusammensetzung von Meta-Gemeinschaften zu bestimmen
- genetische Variationen zu erkennen und die daraus resultierenden Konsequenzen zu untersuchen
Übersicht
Überlegungen vor dem Start eines genomisch-epidemiologischen Analyseprojekts:
- Gibt es ein Referenzgenom?
- Gibt es ein MLST-Profil?
- Sind größere Veränderungen zu erwarten?
- Kann eine erhebliche DNA-Kontamination auftreten - Metacommunity erwartet?
- Gibt es bereits bekannte Resistenz-, Virulenz-, Mykotoxin-Gene, die für den zu analysierenden Stamm spezifisch sind?
Lassen Sie sich von uns beraten - vom Design bis zur Analyse
Beispielprojekte mit genomischer Epidemiologie-Analyse:
- Bestimmen Sie die Pathogenität von bakteriellen DNA-Isolaten mittels MLST
- SNVs und InDels erkennen
- Prüfen Sie die molekulare Epidemiologie von mutmaßlichen Trägerstämmen auf Virulenz, Mykotoxizitätsfaktoren und Arzneimittelresistenz
Anwendungen im Zusammenhang mit der genomischen Epidemiologie-Analyse:
- Amplicon/shotgun metagenomics
- De novo sequencing
- Microbial resequencing
- Shotgun Transcriptomics
Workflow
Ein typischer Workflow für ein genomisch-epidemiologisches Analyseprojekt ist in der folgenden Grafik dargestellt. Bitte beachten Sie, dass unsere hochmodularen Prozesse Ihnen verschiedene Ein- und Ausstiegsmöglichkeiten bieten. Sie können Ihr gesamtes NGS-Projekt oder nur Teile davon an Microsynth auslagern.

Weitere Informationen sowie eine detaillierte technische Beschreibung finden Sie in unserer Application Note Genomic Epidemiology Analysis (siehe Downloads auf der rechten Seite).
Resultate
Bei genomischen Epidemiologiestudien wird in der Regel ein Bakterienstamm sequenziert und auf verschiedene Weise auf seine Pathogenität hin analysiert. Unser genomisch-epidemiologisches Analysemodul hilft Ihnen z. B. bei der Beantwortung folgender Fragen:
- Wie verhält sich der sequenzierte Stamm zu dem bereits bekannten phylogenetischen Baum des analysierten Mikroorganismus? (siehe Tabelle 1)
- Welche Resistenz-, Virulenz- und Mykotoxin-Gene sind in der analysierten Probe vorhanden?
- Gibt es Variationen in Form von Einzelnukleotid-Variationen (SNVs) oder kleinen Insertionen und Deletionen (InDels) im Vergleich zu einem Referenzgenom oder Referenzgenen und was ist deren Konsequenz? (siehe Tabelle 2)
- Wie ist die Zusammensetzung der Meta-Community, falls vorhanden? (siehe Tabelle 3)
- Gibt es neue Gene, die eine signifikante Homologie zu bekannten Proteinfamilien aufweisen? (siehe Tabelle 4)
Tabelle 1. Ergebnis der Multi Locus Sequence Typing (MLST), das den Sequenztyp der in der Probe gefundenen Spezies anzeigt. In diesem Fall umfasste das für die Typisierung verwendete Schema 7 Gene (arcC, aroE, glpF, gmk, pta, tpi und yqiL) zur Identifizierung des jeweiligen Sequenztyps (ST) und des klonalen Komplexes.

Tabelle 2: Zusammenfassende Tabelle der Anzahl der beobachteten SNVs und kleinen InDels in der analysierten Stichprobe einschließlich der Art der Mutation (silent und non-silent).

Tabelle 3. Dieser Ausschnitt eines Ergebnisses einer Shotgun-Metagenomics-Taxonomiezuordnung, zeigt die Zusammensetzung der in der analysierten Probe gefundenen Bakteriengemeinschaft. In diesem Fall enthält die Probe 96 % der Gattung Staphylococcus (Tax Level: G) und 92 % werden als Staphylococcus aureus-Spezies (Tax Level: S) identifiziert.
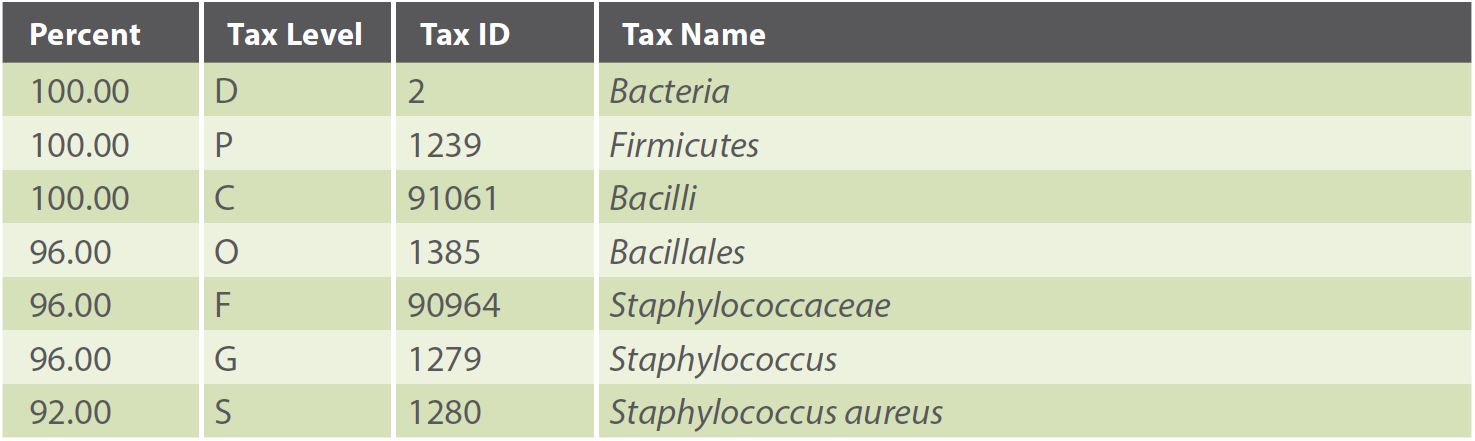
Tabelle 4. Ausschnitt aus einer Tabelle mit den gefundenen homologen Proteindomänen und deren Bedeutung für die vorhergesagten Gene der analysierten Probe.

Bearbeitungszeiten
- Lieferung der Daten innerhalb von 20 Arbeitstagen nach Probeneingang (inklusive Library Erstellung und Sequenzierung)
- Weitere 5 Arbeitstage für die Datenanalyse (Bioinformatik)
- Express-Service auf Anfrage möglich