

Back to top
Whole Genome Sequencing (WGS)

- einzelne Nukleotide bis hin zu großen strukturellen Variationen in Ihrem Modellorganismus zu erkennen
- Off-Target-Mutationen Ihres CRISPR/Cas9-Gene Editing-Experiments zu erkennen
- Ihren bakteriellen Produktionsstamm zu validieren
- mikrobielle Isolate durch MLST zu charakterisieren
Übersicht
Überlegungen vor dem Start eines Whole Genome Sequenzier-Projekts:
- Wissenschaftliches Ziel
- Verfügbarkeit und Qualität eines Referenzgenoms
- SNV-Nachweis und/oder SV-Nachweis
- Optimale Abdeckung/Sequenzierungstiefe und Leselänge
- Besteht der Verdacht auf erhebliche DNA-Kontaminationen
Lassen Sie sich von uns beraten - vom Design bis zur Analyse
Beispielprojekte mit Whole Genome Sequenzierung:
- Detektion von Mutationen, die Krebszellen erworben haben - von SNPs bis zu großen strukturellen Variationen
- Erkennung von Insertionsstellen
- Mutationsverifizierung, Ausschluss von Off-Target-Mutationen
- Genetische Veränderungen in Zuchtstudien
Anwendungen im Zusammenhang mit Whole Genome Sequenzierung:
- RNA Sequencing
- Whole exome resequencing
Workflow
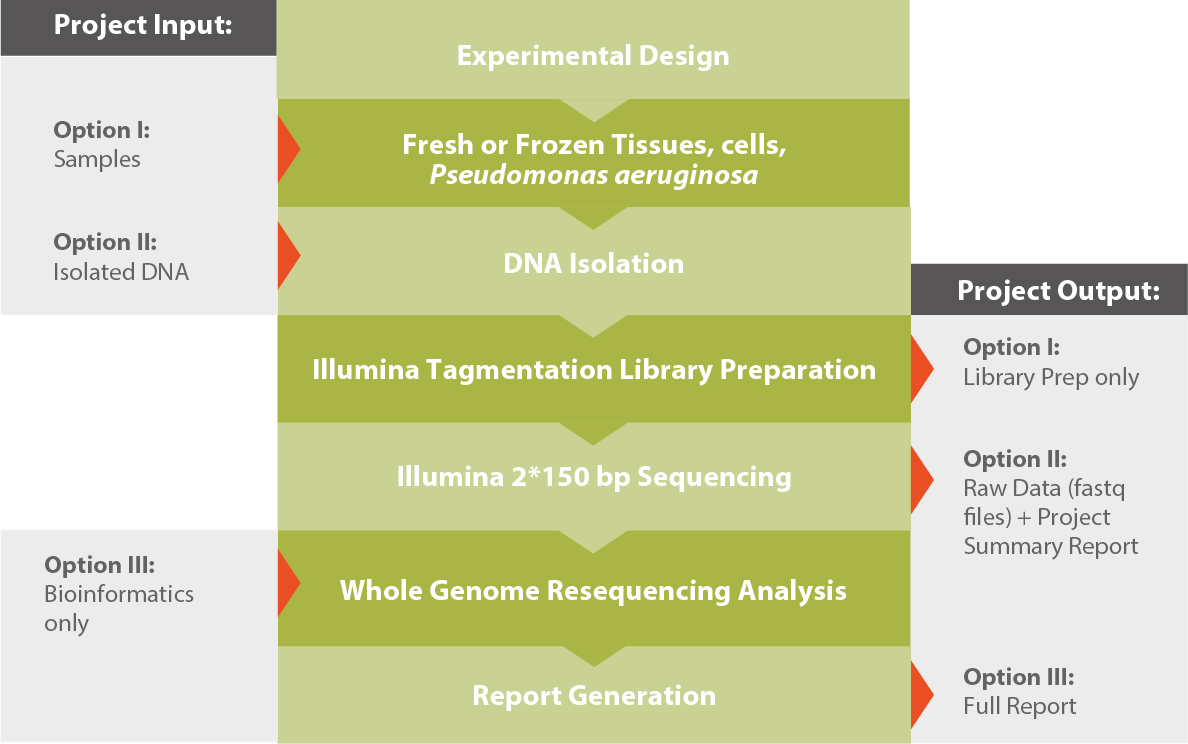
Resultate
Ohne Bioinformatik
Rohdaten:
Wenn kein Analysemodul bestellt wird, liefert Microsynth für Whole Genome Sequencing die unten aufgeführten Schlüsselergebnisse:
- Bewertung der Quantität und Qualität der Sequenzierung (im .xlsx-Format)
Die Bewertung der Quantität und Qualität der Sequenzierungsdaten. - Rohdaten (pro Probe, im .fastq-Format)
Mit den Rohdaten können Sie Ihre eigene Analyse durchführen oder jedes sequenzierte Nukleotid zurückverfolgen. - Ein zusammenfassender Projektbericht (.pdf-Format)
Der Bericht enthält eine Zusammenfassung der wichtigsten Parameter des Projekts.
Mit Bioinformatik
Standard-Bioinformatik-Analyse:
Für unsere Resequenzierungsanwendung bietet das Analysemodul von Microsynth zusätzlich zu den Rohdaten eine Vielzahl von Einblicken, um Ihre wissenschaftlichen Ziele zu erreichen:
- Auswertung der Quantität und Qualität der Sequenzierung (im .xlsx- und .html-Format)
Eine umfassende Bewertung der Quantität und Qualität der Sequenzierung, die wichtige Erkenntnisse liefert. - Alignment/Map-Dateien und Indizes (in den Formaten .bam und .bai)
Zugriff auf Alignment- und Map-Dateien mit den dazugehörigen Indizes für Ihren Komfort. - Analyse der Genomabdeckung und Lesetiefe (in den Formaten .tsv und .bed; siehe Abbildung 1)
Gewinnen Sie Einblicke in die Genomabdeckung und Lesetiefe anhand der im .tsv- und .bed-Format bereitgestellten Ergebnisse. - Strukturelle Variationen & Kopienzahlanalyse (vgl. Tabelle 1 & Abbildung 2)
- Variant Calling von SNVs und kleinen InDels (im .vcf-Format, siehe Tabelle 2 und Tabelle 3 )
Identifizierung von Einzelnukleotidvarianten (SNVs) und kleinen Insertionen/Deletionen (InDels < 50 bp) mit den im .vcf-Format verfügbaren Ergebnissen der Variantenerkennung. - Annotation von Variationen mit Konsequenzen auf Aminosäureebene (im .html-Format, siehe Tabelle 4)
Verstehen Sie Variationen mit potenziellen Konsequenzen auf Aminosäureebene, dargestellt im .html-Format (zugänglich für die meisten Modellorganismen / wenn das Referenzgenom entsprechend annotiert ist).
Optional:
- Filterung von Varianten gegen eine Hintergrundprobe (im .html-Format)
Passen Sie Ihre Analyse an, indem Sie Varianten gegen eine Hintergrundprobe filtern, wobei die Ergebnisse im .html-Format bereitgestellt werden. - Proben-Konsensus-Sequenz (im .fasta- und .gb-Format)
Erhalten Sie eine Proben-Konsensus-Sequenz, die möglicherweise zu einer chimären Sequenz führt, im .fasta- und .gb-Format.
Ergänzende bioinformatische Analyse (gegen Aufpreis)
Genomische Epidemiologie (nur für Prokaryoten):
- Screening auf Übereinstimmungen mit Resistenz- und Virulenzdatenbanken und Mykotoxin-Genen (im .tsv-Format)
Identifizierung von Übereinstimmungen mit Resistenz- und Virulenzdatenbanken mit Bereitstellung der Screening-Ergebnisse im .tsv-Format. - Phylogenetische Typisierung von Probenstämmen (im .pdf-Format)
Verstehen Sie die phylogenetische Verwandtschaft Ihres Probenstamms anhand eines übersichtlichen .pdf-Berichts.
Diese Ergebnisse geben Ihnen einen umfassenden Überblick über Ihre Re-Sequenzierungsanalyse und ermöglichen es Ihnen, aussagekräftige Erkenntnisse zu gewinnen und fundierte Entscheidungen zu treffen.

Tabelle 1: Dieser Ausschnitt aus einer Ergebnistabelle zeigt die entdeckten Einzelnukleotidvariationen, kleine Insertionen und Deletionen und deren Annotation.
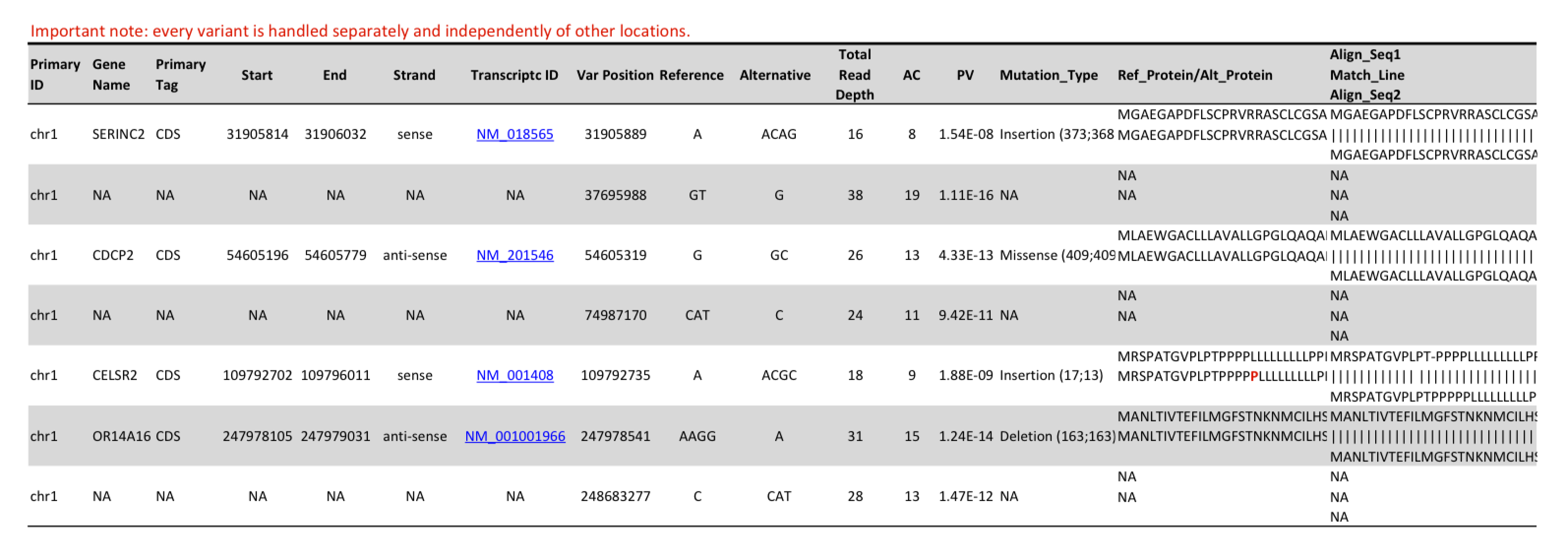

Tabelle 3: Dieser Ausschnitt aus einer Ergebnistabelle listet mutmaßliche Strukturvariationen auf.
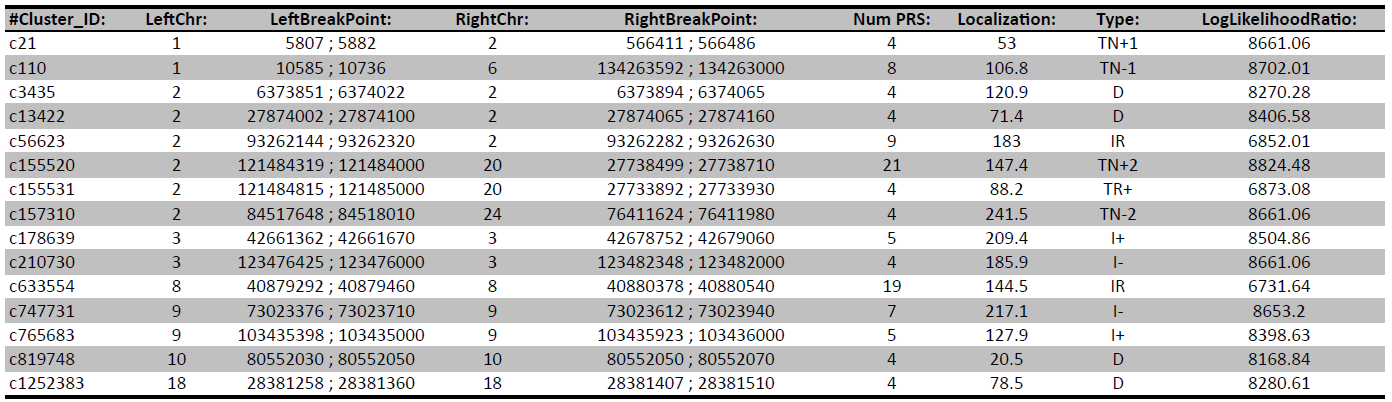
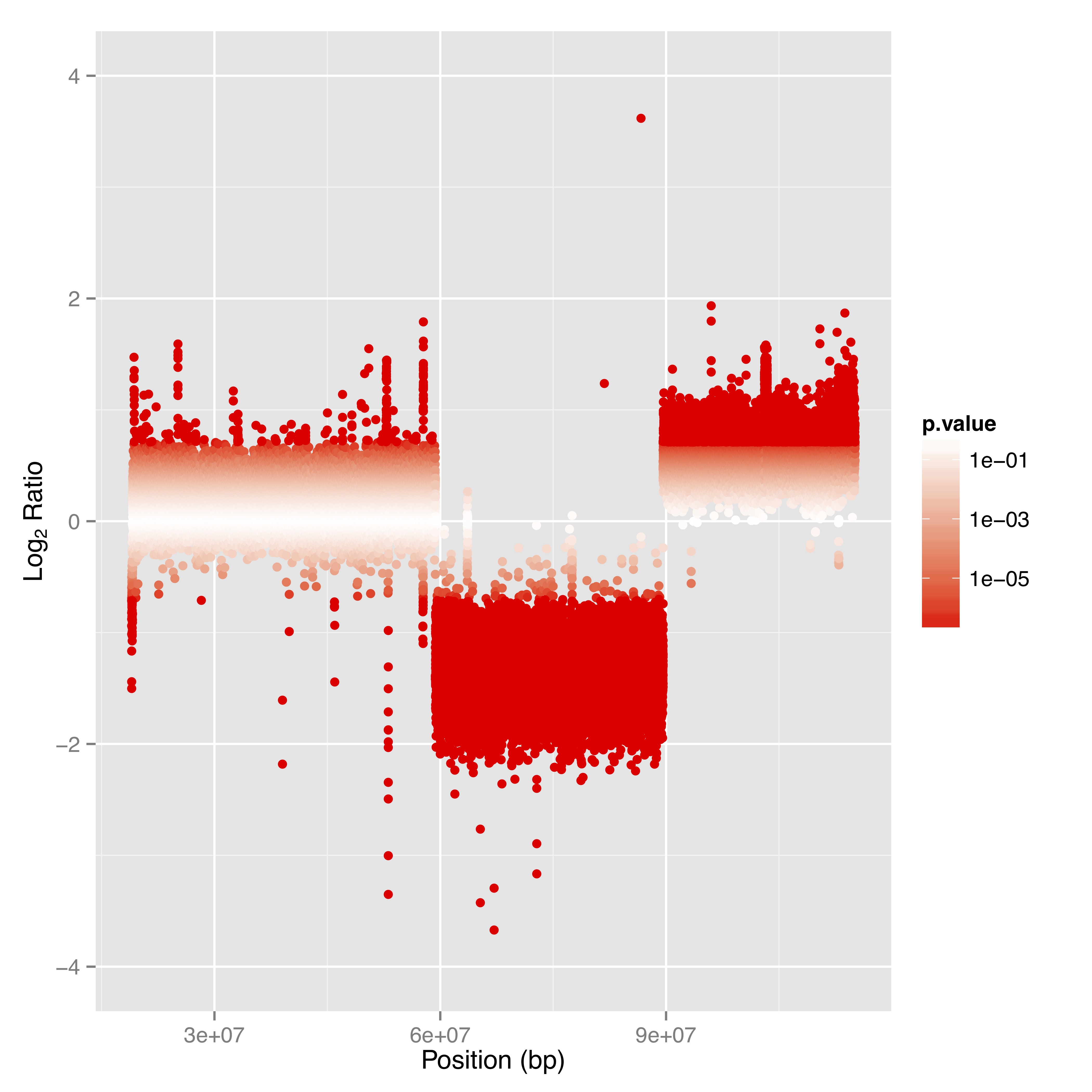
Abbildung 2: Diese Abbildung zeigt eine mögliche Variation der Kopienzahl im Vergleich zu einer Referenzprobe.
Tabelle 4: Tabelle mit detaillierten Informationen zu jeder beobachteten Variation, wie z. B. Typ und Position der Variante, betroffenes Protein, einschließlich Referenz- und veränderter Sequenz.

Bearbeitungszeiten
- Lieferung der Daten innerhalb von 20 Arbeitstagen nach Probeneingang (inklusive Library Erstellung und Sequenzierung)
- Weitere 10 Arbeitstage für die Datenanalyse (Bioinformatik)
- Express-Service auf Anfrage möglich