

Back to top
Whole Genome Metagenomics Analyse der Mikrobiota

- das genetische Potenzial Ihrer bakteriellen Gemeinschaftsproben zu untersuchen
- eine hypothesenfreie taxonomische Analyse zu erstellen
Übersicht
Überlegungen vor dem Start eines Shotgun-Metagenomik-Projekts:
- Probenmengen und Herkunft?
- Sequenziertiefe (Sensitivität)?
- Fokus auf genetisches Potenzial oder auf Taxonomie?
- Komplexität und Zusammensetzung der Artengemeinschaft?
- Deskriptive und/oder empirische Studie?
Lassen Sie sich von uns beraten - vom Design bis zur Analyse
Beispielprojekte mit Shotgun-Metagenomik:
- Community-Analyse von mikrobiellen Gemeinschaften
- Verschiebung von Gemeinschaften und genomischem Potential bei veränderten Umweltbedingungen
- Evolutionäre Anpassungen in Mikro-Umgebungen
- Analyse auf Gen-Ebene von nicht kultivierbaren mikrobiellen Gemeinschaften
- Virusnachweis in Säugetiergeweben und Exsudaten
Anwendungen im Zusammenhang mit Shotgun-Metagenomik:
- Amplicon metagenomics
- Shotgun metatranscriptomics
- Bacterial resequencing
Workflow
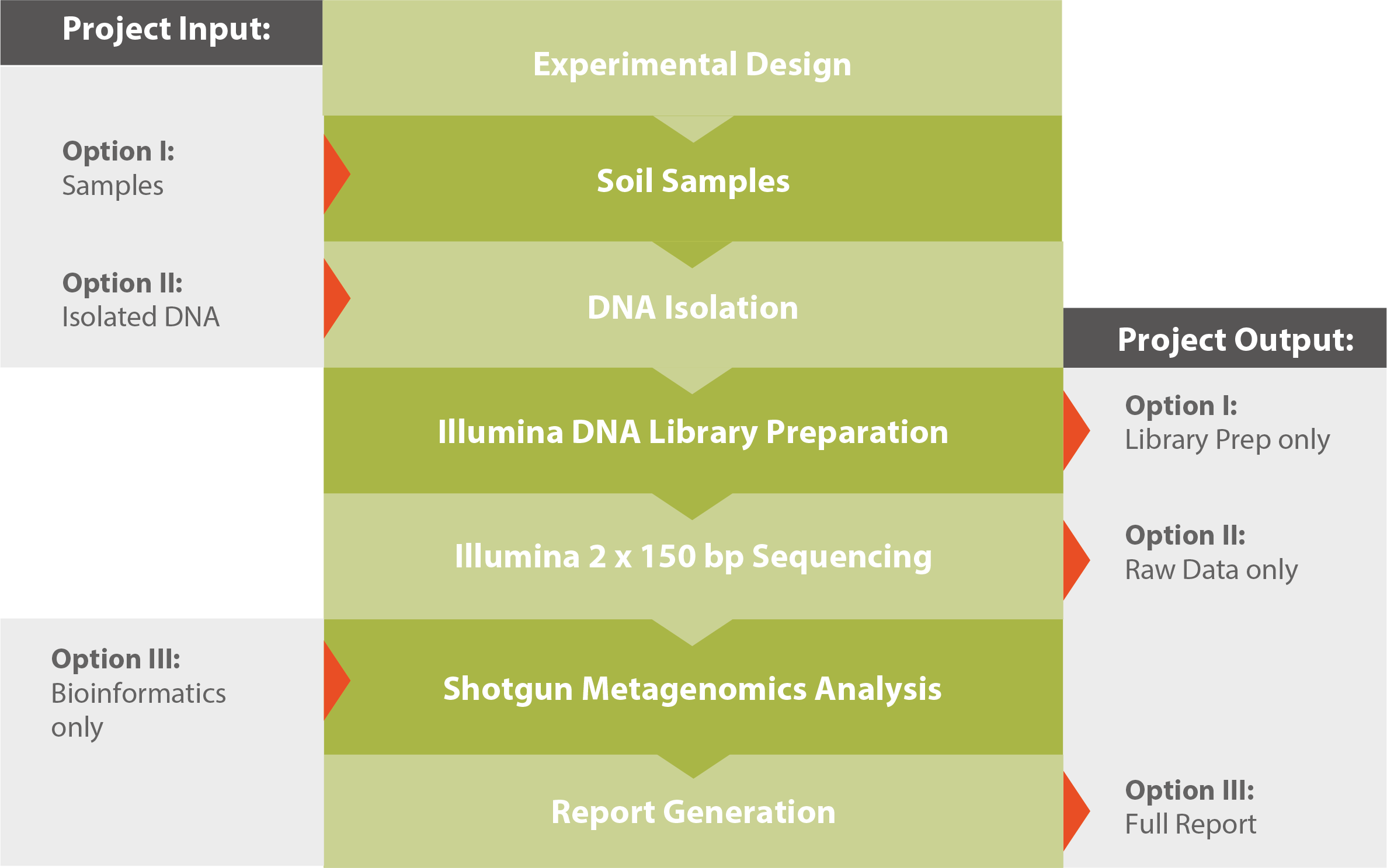
Weitere Informationen sowie eine detaillierte technische Beschreibung findenSie in unserer Application Note Shotgun Metagenomics (siehe Downloads auf der rechten Seite).
Resultate
Im Gegensatz zu amplikonbasierten Metagenomikstudien beruht die Shotgun-Metagenomik nicht auf einem einzelnen phylogenetischen Markergen, sondern berücksichtigt die gesamte aus einer Probe gewonnene DNA. Daher können eukaryotische und prokaryotische Organismen in einer mikrobiellen Gemeinschaft gleichzeitig analysiert werden. Shotgun-Metagenomik beschränkt sich nicht auf die taxonomische Zusammensetzung einer Gemeinschaft, sondern offenbart auch deren Inventar an funktionellen Genen.
Die Analyse von Shotgun-Metagenomik-Datensätzen ist aufgrund ihrer Größe eine Herausforderung. Insbesondere das Alignment der Reads gegen eine Referenzdatenbank ist der größte Engpass und erfordert entsprechende Hard- und Software-Ressourcen. Unser Shotgun-Metagenomics Analysemodul läuft auf Hochleistungsservern unter Verwendung der schnellsten verfügbaren Tools. Das Modul hilft Ihnen, die folgenden Fragen zu beantworten:
- Wie ist die taxonomische Zusammensetzung (sowohl eukaryotisch als auch prokaryotisch) der mikrobiellen Gemeinschaft? (siehe Abbildung 1)
- Wie groß ist das funktionelle Potenzial der mikrobiellen Gemeinschaft? (siehe Abbildung 2)
- Gibt es taxonomische und funktionelle Merkmale, die zwischen den Gemeinschaften oder Bedingungen unterschiedlich häufig vorkommen? (siehe Abbildung 3)

Abbildung 1: Wortwolke, die die bakterielle Zusammensetzung einer Gemeinschaft auf Phylum-Ebene darstellt. Die Größe der Wörter ist proportional zur Phylum-Häufigkeit.
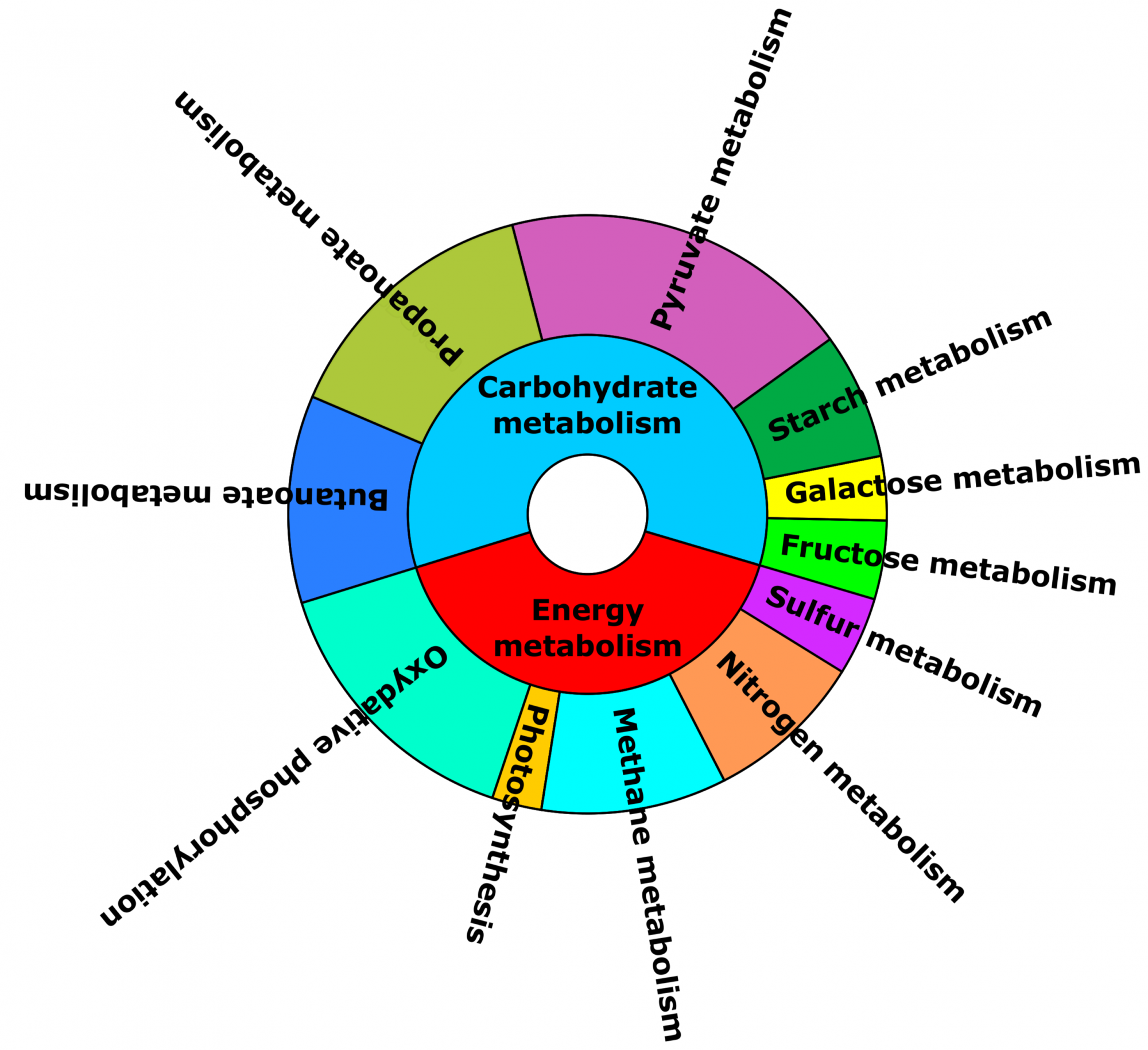
Abbildung 2: Radiales Baumdiagramm, das die KEEG-Funktionen Kohlenhydratstoffwechsel und Energiestoffwechsel und die zugehörigen Untergruppen darstellt.
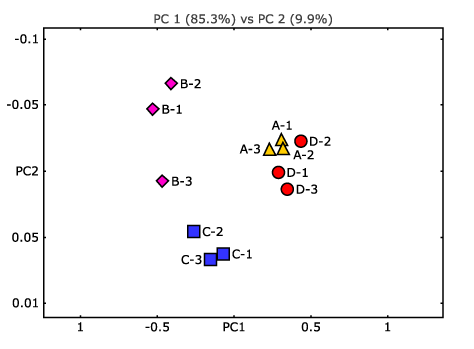
Abbildung 3: PCoA für den Multi-Sample-Vergleich auf Basis von Bray-Curtis-Dissimilaritäten für das funktionelle Potenzial verschiedener Proben auf Basis der funktionellen InterPro-Annotation.
Bearbeitungszeiten
- Lieferung der Daten innerhalb von 25 Arbeitstagen nach Probeneingang (einschließlich Library Erstellung und Sequenzierung)
- Weitere 10 Arbeitstage für die Datenanalyse (Bioinformatik)
- Express-Service auf Anfrage möglich