

Back to top
mRNA-Sequenzierung für die differentielle Genexpressionsanalyse

- die unter unterschiedlichen Bedingungen erhaltenen Genexpressionsprofile zu vergleichen
- Genetische Profile von der Transkript- bis zur Ebene der Signalwege zu untersuchen
Übersicht
Überlegungen vor dem Start eines mRNA-Sequenzierungsprojekts:
- Wissenschaftliches Ziel
- Ausgangsmaterial & gewünschte Dienstleistungen (Isolierung bis Bioinformatik)
- Organismus und RNA-Fraktion (Poly(A)-Anreicherung, Ribo-Abreicherung)
- Verfügbare Daten (Modellorganismus, Referenzgenom)
- Sequenzierungstiefe (Empfindlichkeit)
- Replikate (Sicherheit)
- Sequenzierungslänge (Spezifität)
- Probenmengen (Komplexität)
- Versuchsaufbau (Anzahl der Proben, Wiederholungen, zu vergleichende Bedingungen)
Lassen Sie sich von uns beraten - vom Design bis zur Analyse
Beispielprojekte mit mRNA-Sequenzierung:
- Funktionelle Protein- und Signalwegstudien
- Krankheitsbedingte Veränderungen der Genexpression
- Knock-out oder Knock-in Experimente
- Teil einer Omics-Charakterisierung
- Funktionelle Veränderungen aufgrund von Spezies-Interaktionen
- Entdeckung von neuen Genen oder nicht-kodierender regulatorischer RNA
- Erkennung von RNA-Varianten
- Medikamenten-Tests
Anwendungen im Zusammenhang mit der mRNA-Sequenzierung:
- Shotgun metatranscriptomics
- Small RNA sequencing
Workflow

Weitere Informationen sowie eine detaillierte technische Beschreibung finden Sie in unserer Application Note Illumina RNA Sequencing (siehe Downloads auf der rechten Seite).
Resultate
Ohne Bioinformatik
Rohdaten
Wenn kein bioinformatisches Analysemodul bestellt wird, liefert Microsynth für die mRNA-Sequenzierung die unten aufgeführten wichtigsten Ergebnisse:
- Bewertung der Quantität und Qualität der Sequenzierung (im .xlsx-Format)
Bewerten Sie die Quantität und Qualität der Sequenzierungsdaten. - Rohdaten (pro Probe, im .fastq-Format)
Zugriff auf Rohdaten für individuelle Analysen oder als Referenz. - Ein zusammenfassender Projektbericht (.pdf-Format)
Fassen Sie die wichtigsten Projektparameter zusammen.
Mit Bioinformatik
Standard-Bioinformatik-Analyse
Für unsere mRNA-Sequenzierungsanwendung liefert das Microsynth-Modul eine Vielzahl von Erkenntnissen, die auf Ihre wissenschaftlichen Ziele ausgerichtet sind:
Expressionsanalyse:
- Umfassender Bericht (interaktives .html-Format)
Tauchen Sie tiefer in die Daten ein, sortieren und filtern Sie die Ergebnisse interaktiv. - Alignment/Map-Dateien und Indizes (im .bam- und .bai-Format)
Greifen Sie auf Alignment-/Map-Dateien zusammen mit den entsprechenden Indizes zu, um jeden einzelnen Read bis zum Referenzgenom zu verfolgen. - Read Counts zu annotierten Merkmalen (im .tsv-Format)
Erhalten Sie Read-Zahlen, die annotierten Merkmalen wie Genen oder Transkripten zugeordnet sind, um eine detaillierte quantitative Analyse durchzuführen. - Visualisierung der statistischen Analyse (z. B. im .png-Format, siehe Abbildung 1-5)
Greifen Sie auf visuell informative Darstellungen von statistischen Analysen zu, einschließlich Vulkandiagrammen, Heatmaps, angereicherten Pfaden und Exon-Karten.
Exklusiv für eukaryotische Transkriptome:
- Differenzielle Genexpressionsanalyse (im .tsv- und interaktiven .html-Format, siehe Abbildung 3)
Erkunden Sie die differentielle Genexpression mit umfassenden Ergebnissen im Tabellen- und HTML-Format. - Geneset-Analyse (im .tsv- und .html-Format, siehe Abbildung 4)
Entdecken Sie angereicherte Pfade durch die Geneset-Analyse, die im Tabellen- und HTML-Format verfügbar ist. - Differenzielle Exon-Nutzungsanalyse (im .tsv- und .html-Format, siehe Abbildung 5)
Erforschen Sie das alternative Spleißen durch die differenzielle Exon-Nutzungsanalyse, deren Ergebnisse sowohl im Tabellen- als auch im HTML-Format zur Verfügung stehen.
Diese Ergebnisse liefern verwertbare Daten aus Ihrer mRNA-Sequenzierungsanalyse und ermöglichen aufschlussreiche Entscheidungen.
Ergänzende bioinformatische Analysen ( gegen Aufpreis)
- Variant Calling von SNVs und kleinen (<50bp) InDels im Transkriptom (im .vcf-Format)
Identifizierung von Einzelnukleotidvarianten (SNVs) und kleinen Insertionen/Deletionen (InDels) im Transkriptom durch Variantenaufrufe im .vcf-Format.
Bei entsprechendem Versuchsaufbau und der Verfügbarkeit relevanter öffentlicher Daten bieten diese zusätzlichen Dienste ein tieferes Verständnis Ihrer Proben.
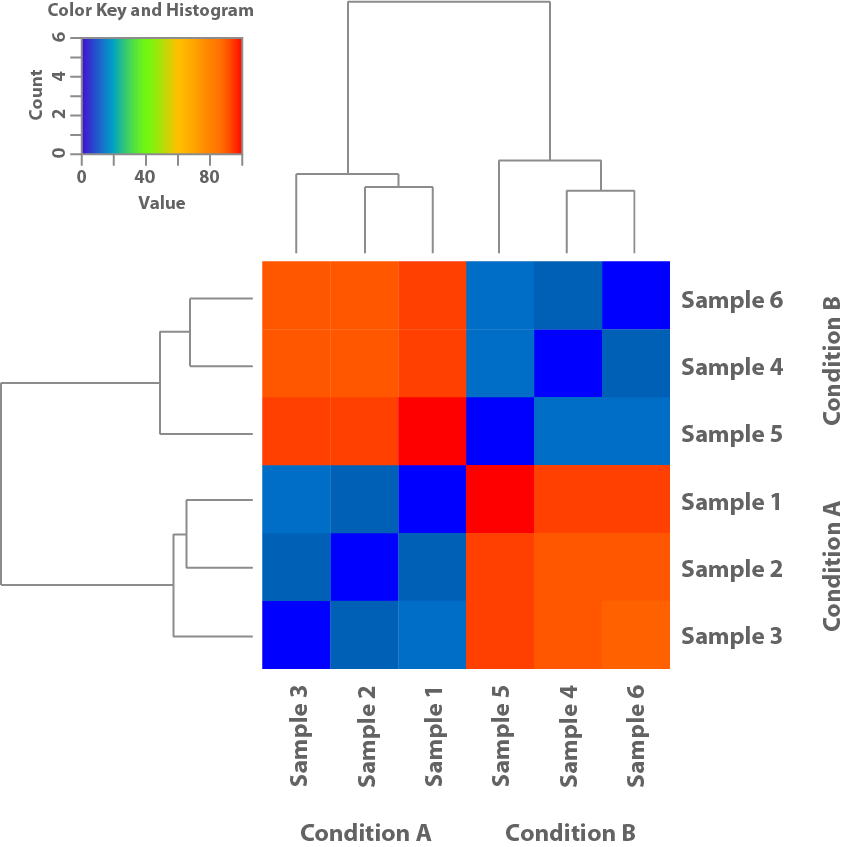
Abbildung 1: Diese Heatmap basiert auf den Expressionsmustern der Proben und zeigt deren Ähnlichkeit zueinander. Dies hilft zu klären, ob die im Experiment verwendeten Bedingungen zu unterschiedlichen Expressionsmustern führen.
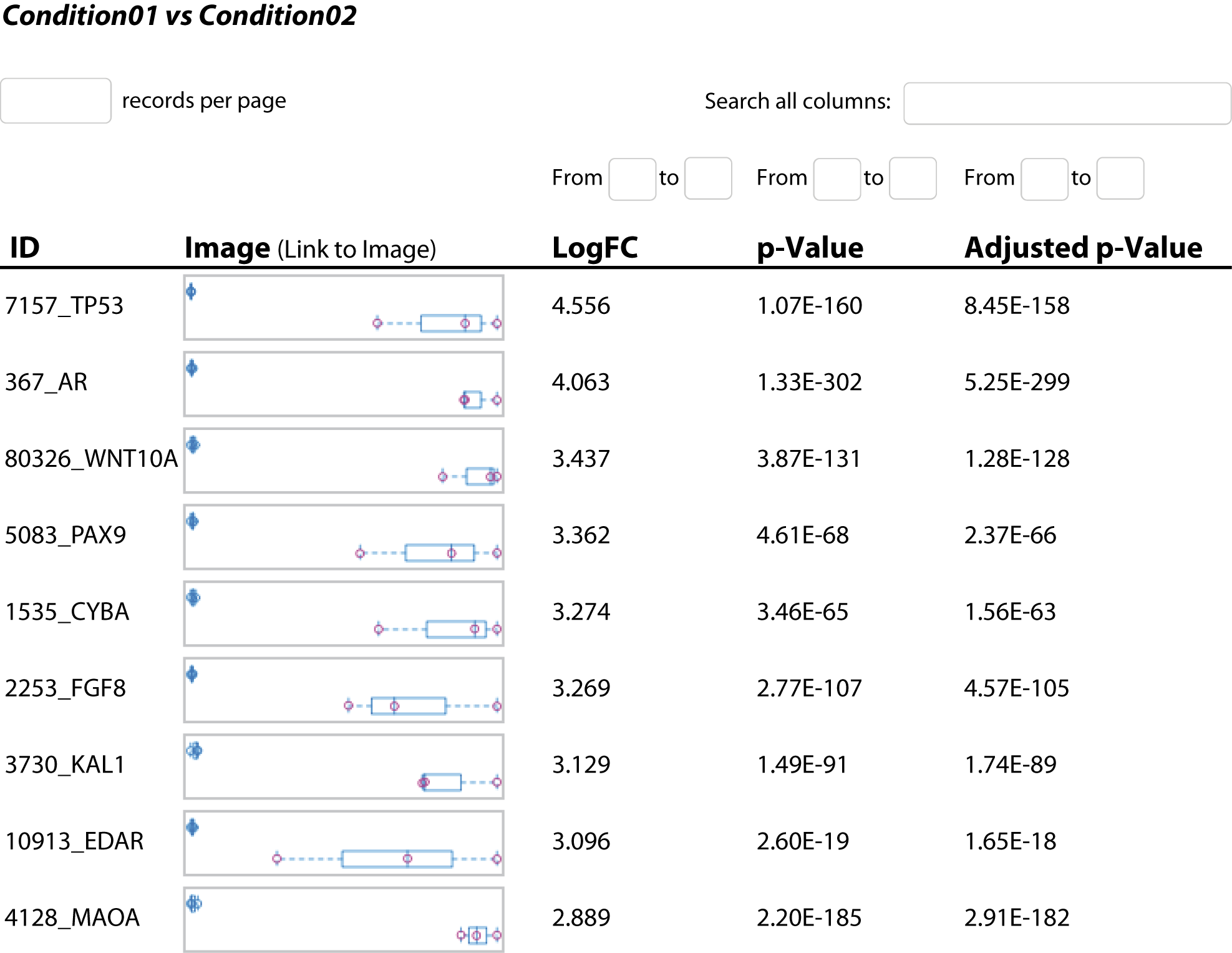
Abbildung 3: Für alle gemessenen Gene sind detaillierte Statistikwerte wie log fold change und deren Signifikanz zur weiteren Untersuchung aufgeführt.

Abbildung 5: Die alternative Splice-Analyse hilft, das wahrscheinlichste Transkript eines Gens zum Zeitpunkt des Experiments zu identifizieren.
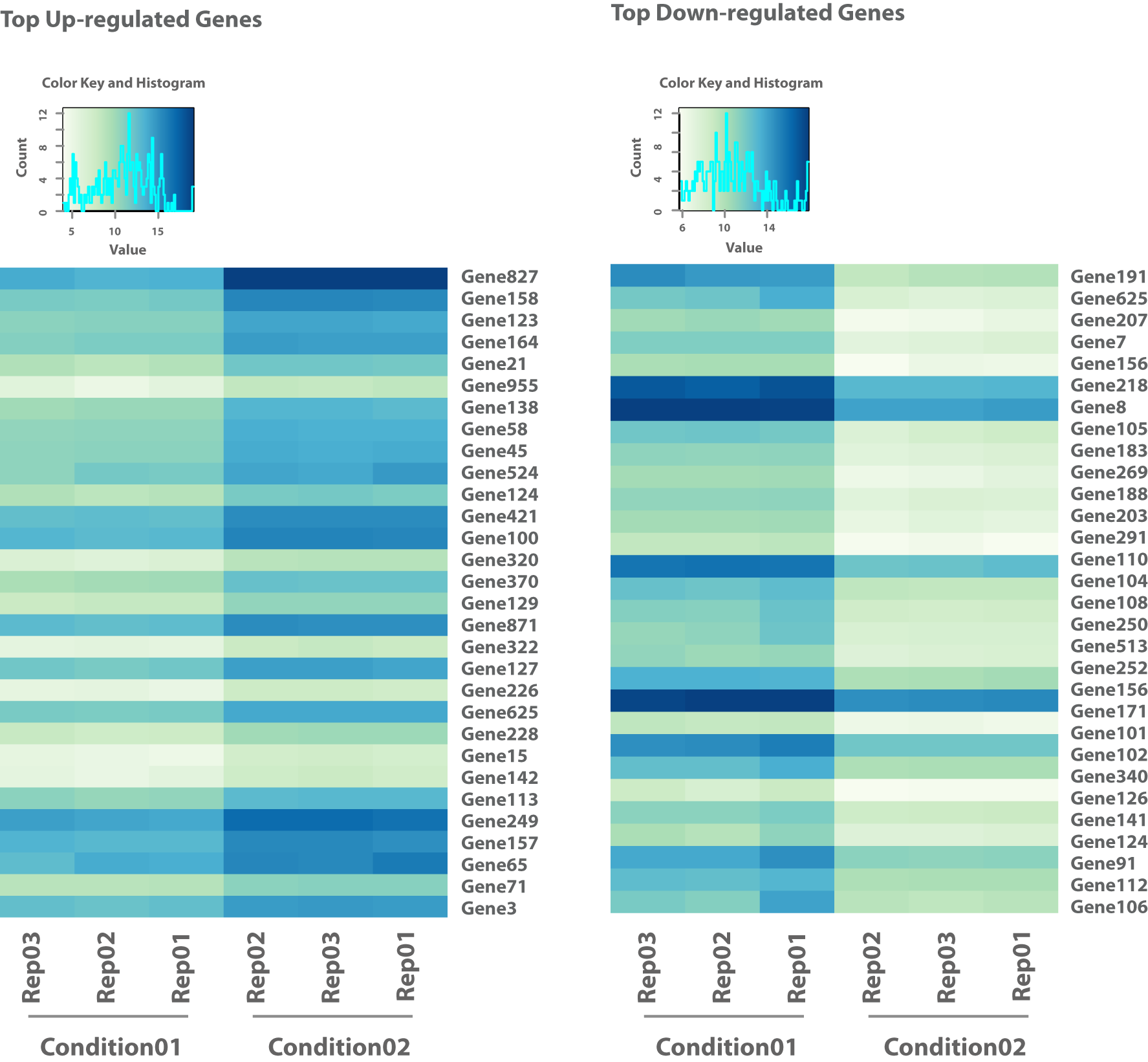
Abbildung 2: Die zweite Heatmap zeigt die top hochregulierten und top herunterregulierten Gene aus einem paarweisen Vergleich zweier Bedingungen (z.B. Stressor vs. Kontrolle).
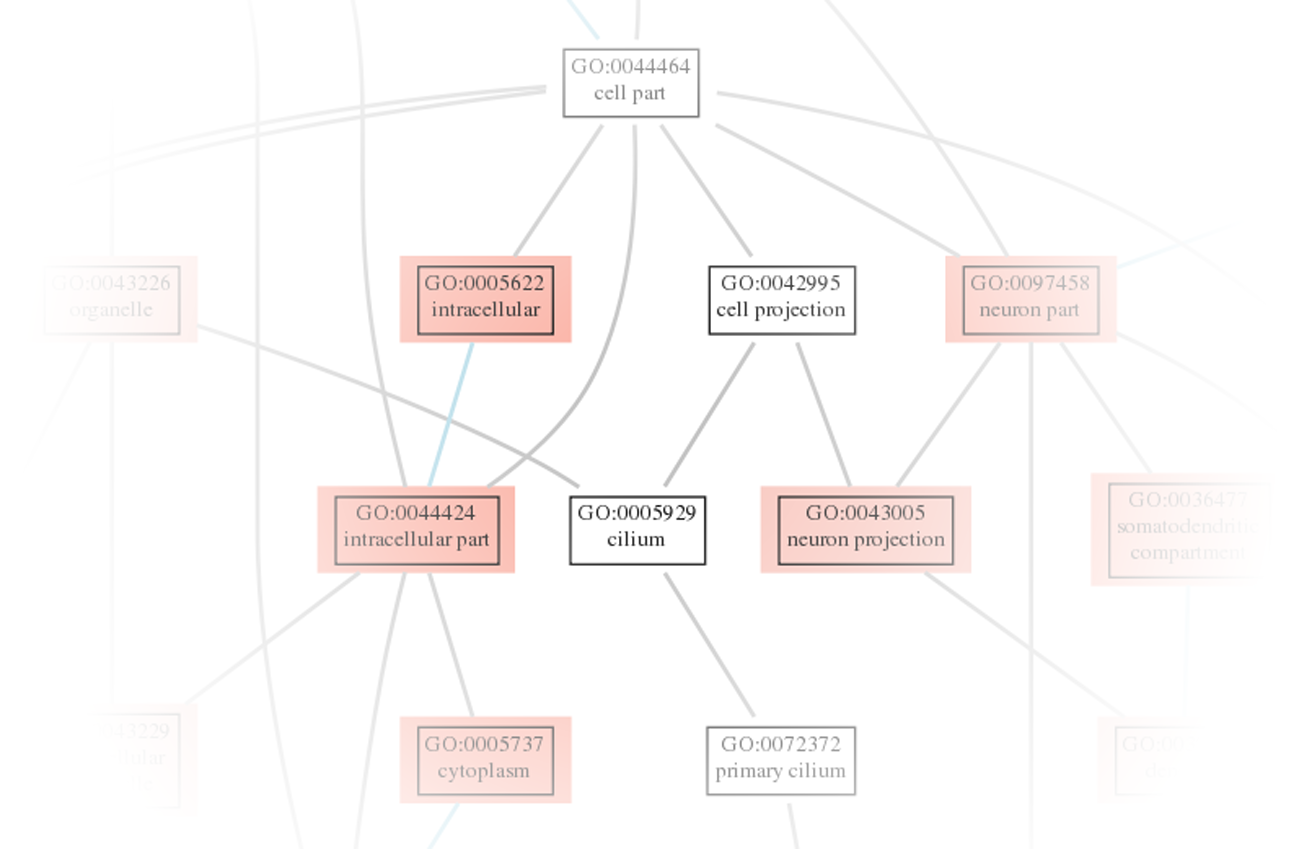
Abbildung 4: Die Signalweganreicherungsanalyse hilft bei der Identifizierung von differentiell regulierten Signalwegen, die wiederum beobachtete Phänotypen erklären können.
Bearbeitungszeiten
- Lieferung der Daten innerhalb von 25 Arbeitstagen nach Probeneingang (einschließlich Library Erstellung und Sequenzierung)
- Zusätzliche 5 Arbeitstage für die Datenanalyse (Bioinformatik)
- Express-Service auf Anfrage möglich